
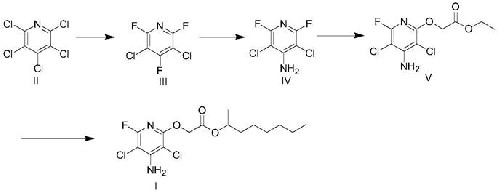
1.本发明涉及有机合成领域,具体涉及一种氯氟吡氧乙酸异辛酯的制备方法。
背景技术:
2.氯氟吡氧乙酸异辛酯(又名氟草烟仲辛酯)是选择性内吸传导型苗后除草剂,主要用于防除小麦、大麦、玉米等禾科作物田中的各种阔叶杂草,目前在国内已获得广泛应用。目前已开发的氯氟吡氧乙酸异辛酯合成方法普遍存在原料难获取、制备成本过高、工艺路线复杂、收率纯度低以及污染严重等问题,因此急需开发一种成本低廉、操作简单、收率纯度高、安全环保的氯氟吡氧乙酸异辛酯制备方法。
3.专利公开了一种氯氟吡氧乙酸异辛酯的制备方法:以4-氨基-3,5-二氯-6-氟-2-吡啶酚钾为原料,先与过量氯乙酸甲酯反应得到4-氨基-3,5-二氯-6-氟-2-吡啶氧乙酸甲酯,再与过量仲辛醇发生酯交换反应,得到目标产物。该合成方法的原料4-氨基-3,5-二氯-6-氟-2-吡啶酚钾难获取,工艺成本过高,且氯乙酸甲酯的毒性较大,不利于氯氟吡氧乙酸异辛酯的工业化生产。
4.专利公开了一种一锅法合成氯氟吡氧乙酸异辛酯的方法:先将仲辛醇与羟基乙酸发生酯化反应,再与4-氨基-3,5-二氯-2,6-二氟吡啶缩合后得到目标产物。该制备方法同样存在起始物料4-氨基-3,5-二氯-2,6-二氟吡啶难获取成本高的问题,且发明人通过还原该合成方法,得到的氯氟吡氧乙酸异辛酯纯度仅为92%,不足以保证产品的质量。
技术实现要素:
5.本发明要解决的技术问题是:克服上述现有技术的不足,提供一种原料易得、成本低廉、安全环保的高纯度氯氟吡氧乙酸异辛酯的制备方法。
6.本发明解决上述技术问题的技术方案如下:
7.本发明提供了一种氯氟吡氧乙酸异辛酯的制备方法,其特征在于,包括如下步骤:
[0008][0009]
(1)式ii化合物在氟化试剂/相转移催化剂存在下生成式iii化合物;
[0010]
(2)式iii化合物与氨试剂反应生成式iv化合物;
[0011]
(3)式iv化合物在强碱试剂存在下,与乙醇酸乙酯反应生成式v化合物;
[0012]
(4)式v化合物在催化剂存在下,与仲辛醇反应生成式i化合物;
[0013]
进一步的,所述步骤(1)中氟化试剂/相转移催化剂为kf/四丁基氟化铵、kf/四丁基氯化铵或均为四甲基氟化铵,优选kf/四丁基氟化铵;
[0014]
进一步的,所述步骤(1)中氟化试剂、相转移催化剂与式ii化合物的摩尔比为5~15:3~8:1,优选10:5:1;
[0015]
进一步的,所述步骤(2)中的氨试剂为液氨或氨水,优选25%氨水;
[0016]
进一步的,所述步骤(2)中反应温度为70℃~90℃,优选80℃;
[0017]
进一步的,所述步骤(3)中强碱试剂为nah、碳酸铯或叔丁基锂中的一种或几种,优选nah;
[0018]
进一步的,所述步骤(3)中强碱试剂、乙醇酸乙酯与式iv化合物的摩尔比为1~3:1~2:1,优选2:1:1;
[0019]
进一步的,所述步骤(3)中反应溶剂为dma、dmf或dmso中的一种或几种,优选dma;所述反应溶剂与式iv化合物的体积/摩尔比为2~5:1。
[0020]
进一步的,所述步骤(4)中催化剂为钛酸丁酯或钛酸乙酯中的一种或几种,优选钛酸丁酯;
[0021]
进一步的,所述步骤(4)中催化剂、仲辛醇与式v化合物的摩尔比为0.15~0.25:2~5:1,优选0.2:4:1。
[0022]
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
[0023]
本发明的有益效果在于:以五氯吡啶为原料制备氯氟吡氧乙酸异辛酯,原料便宜易得;经3,5-二氯-2,6-二氟-4-氨基吡啶直接生成2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯,避免了大量含氯试剂与酚的反应,安全环保;同时反应后处理简单,收率和纯度较高,适合氯氟吡氧乙酸异辛酯的工业化生产。
具体实施方式
[0024]
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
[0025]
实施例1:
[0026][0027]
(1)3,5-二氯-2,4,6-三氟吡啶(式iii)
[0028]
向50ml三颈瓶中加入式ii化合物(2.49g,)、kf(5.81g,)、无水四丁基氟化铵(13.05g,),加入无水dma(20ml),氮气保护下加热至100℃反应2h。反应液冷却至室温,倒入冰水中,用ea(30ml
×
3)萃取,合并有机相用水(30ml
×
2)洗,有机相用无水硫酸钠干燥,浓缩得淡黄色油状物2.02g,纯度98.2%,收率98.0%。esi-ms(m/z):201.94
[m+h]
。
[0029]
(2)3,5-二氯-2,6-二氟-4-氨基吡啶(式iv)
[0030]
向25ml三颈瓶中加入25%氨水(10ml),冷却至0℃,缓慢加入3,5-二氯-2,4,6-三氟吡啶(1.5g,7.),升温至80℃反应2.5h,冷却至室温,过滤得白色滤饼,少量水洗,干燥得白色固体1.44g,纯度98.2%,收率95.6%。m.p.111-113℃,esi-ms(m/z):198.96[m+h]
。
[0031]
(3)2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(式v)
[0032]
向50ml三颈瓶中加入氢化钠(315mg,13.)和dma(10ml),氮气保护,缓慢滴加乙醇酸乙酯(684mg,6.)的dma(5ml)溶液,室温反应0.5h,加入3,5-二氯-2,6-二氟-4-氨基吡啶(1.3g,6.)的dma(5ml)溶液,加热至内温35℃反应1.5小时。反应液倒入冰水中,dcm(50ml
×
3)萃取,分液,无水硫酸钠干燥,浓缩得无色油状物1.94g,纯度95.1%,收率99.0%。esi-ms(m/z):283.01[m+h]
。
[0033]
(4)氯氟吡氧乙酸异辛酯(式i)
[0034]
向50ml三颈瓶中加入2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(1.6g,5.),仲辛醇(2.94g,22.6mmol),钛酸丁酯(394mg,1.)和甲苯(20ml),加热至内温110℃,反应5小时。冷却至室温,加dcm(100ml)稀释反应液,另加水(20ml
×
3)洗涤有机相,分液,无水硫酸钠干燥,浓缩得淡黄色产物2.05g,收率97.5%,hplc纯度99.2%。esi-ms(m/z):367.10[m+h]
。
[0035]1h-nmr(,)δ:6.802(s,2h);5.124-4.956(m,1h);4.915(s,2h);1.667-1.519(m,2h);1.328-1.296(m,8h);1.226-1.195(d,3h);0.923-0.884(t,3h)。
[0036]
实施例2:
[0037]
(1)3,5-二氯-2,4,6-三氟吡啶(式iii)
[0038]
向50ml三颈瓶中加入式ii化合物(2.49g,)、kf(5.81g,)、无水四丁基氯化铵(13.90g,),加入无水dma(20ml),氮气保护下加热至100℃反应2h。反应液冷却至室温,倒入冰水中,用ea(30ml
×
3)萃取,合并有机相用水(30ml
×
2)洗,有机相用无水硫酸钠干燥,浓缩得淡黄色油状物1.94g,纯度97.5%,收率93.8%。esi-ms(m/z):201.94[m+h]
。
[0039]
(2)3,5-二氯-2,6-二氟-4-氨基吡啶(式iv)
[0040]
向25ml三颈瓶中加入50%氨水(10ml),冷却至0℃,缓慢加入3,5-二氯-2,4,6-三氟吡啶(1.5g,7.),升温至70℃反应2.5h,冷却至室温,过滤得白色滤饼,少量水洗,干燥得白色固体1.45g,纯度98.1%,收率96.0%。m.p.111-113℃,esi-ms(m/z):198.96[m+h]
。
[0041]
(3)2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(式v)
[0042]
向50ml三颈瓶中加入氢化钠(315mg,13.)和dma(10ml),氮气保护,缓慢滴加乙醇酸乙酯(718mg,6.)的dma(5ml)溶液,室温反应0.5h,加入3,5-二氯-2,6-二氟-4-氨基吡啶(1.3g,6.)的dma(5ml)溶液,加热至内温50℃反应1.5小时。反应液倒入冰水中,dcm(50ml
×
3)萃取,分液,无水硫酸钠干燥,浓缩得无色油状物1.66g,纯度96.6%,收率98.5%。esi-ms(m/z):283.01[m+h]
。
[0043]
(4)氯氟吡氧乙酸异辛酯(式i)
[0044]
向50ml三颈瓶中加入2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(1.6g,5.),仲辛醇(2.94g,22.6mmol),钛酸丁酯(394mg,1.)和甲苯(20ml),加热至内温110℃,反应5小时。冷却至室温,加dcm(100ml)稀释反应液,另加水(20ml
×
3)洗涤有机相,分液,无水硫酸钠干燥,浓缩得淡黄色产物2.04g,收率96.9%,hplc纯度98.8%。esi-ms(m/z):367.10[m+h]
。
[0045]
实施例3:
[0046]
(1)3,5-二氯-2,4,6-三氟吡啶(式iii)
[0047]
向50ml三颈瓶中加入式ii化合物(2.49g,)、kf(9.28g,),加入无水dma(20ml),氮气保护下加热至100℃反应2h。反应液冷却至室温,倒入冰水中,用ea(30ml
×
3)萃取,合并有机相用水(30ml
×
2)洗,有机相用无水硫酸钠干燥,浓缩得淡黄色油状物1.87g,纯度97.7%,收率90.5%。esi-ms(m/z):201.94[m+h]
。
[0048]
(2)3,5-二氯-2,6-二氟-4-氨基吡啶(式iv)
[0049]
向25ml三颈瓶中加入20%氨水(10ml),冷却至0℃,缓慢加入3,5-二氯-2,4,6-三氟吡啶(1.5g,7.),升温至90℃反应2.5h,冷却至室温,过滤得白色滤饼,少量水洗,干燥得白色固体1.45g,纯度97.5%,收率95.5%。m.p.111-113℃,esi-ms(m/z):198.96[m+h]
。
[0050]
(3)2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(式v)
[0051]
向50ml三颈瓶中加入碳酸铯(4.28g,13.)和dma(10ml),氮气保护,缓慢滴加乙醇酸乙酯(684mg,6.)的dma(5ml)溶液,室温反应0.5h,加入3,5-二氯-2,6-二氟-4-氨基吡啶(1.3g,6.)的dma(5ml)溶液,加热至内温35℃反应1.5小时。反应液倒入冰水中,dcm(50ml
×
3)萃取,分液,无水硫酸钠干燥,浓缩得无色油状物1.76g,纯度96.0%,收率90.8%。esi-ms(m/z):283.01[m+h]
。
[0052]
(4)氯氟吡氧乙酸异辛酯(式i)
[0053]
向50ml三颈瓶中加入2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(1.6g,5.),仲辛醇(2.94g,22.6mmol),钛酸乙酯(258mg,1.)和甲苯(20ml),加热至内温110℃,反应5小时。冷却至室温,加dcm(100ml)稀释反应液,另加水(20ml
×
3)洗涤有机相,分液,无水硫酸钠干燥,浓缩得淡黄色产物1.83g,收率88.0%,hplc纯度98.2%。esi-ms(m/z):367.10[m+h]
。
[0054]
实施例4:
[0055]
(1)3,5-二氯-2,4,6-三氟吡啶(式iii)
[0056]
向50ml三颈瓶中加入式ii化合物(2.49g,)、kf(5.81g,)、无水四丁基氟化铵(20.91g,),加入无水dma(20ml),氮气保护下加热至100℃反应2h。反应液冷却至室温,倒入冰水中,用ea(30ml
×
3)萃取,合并有机相用水(30ml
×
2)洗,有机相用无水硫酸钠干燥,浓缩得淡黄色油状物2.01g,纯度98.2%,收率97.7%。esi-ms(m/z):201.94[m+h]
。
[0057]
(2)3,5-二氯-2,6-二氟-4-氨基吡啶(式iv)
[0058]
向25ml三颈瓶中加入25%氨水(10ml),冷却至0℃,缓慢加入3,5-二氯-2,4,6-三氟吡啶(1.5g,7.),升温至105℃反应2.5h,冷却至室温,过滤得白色滤饼,少量水洗,干燥得白色固体1.39g,纯度96.6%,收率90.2%。m.p.111-113℃,esi-ms(m/z):198.96[m+
h]
。
[0059]
(3)2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(式v)
[0060]
向50ml三颈瓶中加入碳酸钾(2.72g,19.7mmol)和dma(10ml),氮气保护,缓慢滴加乙醇酸乙酯(684mg,6.)的dma(5ml)溶液,室温反应0.5h,加入3,5-二氯-2,6-二氟-4-氨基吡啶(1.3g,6.)的dma(5ml)溶液,加热至内温35℃反应1.5小时。反应液倒入冰水中,dcm(50ml
×
3)萃取,分液,无水硫酸钠干燥,浓缩得无色油状物1.70g,纯度94.9%,收率86.6%。esi-ms(m/z):283.01[m+h]
。
[0061]
(4)氯氟吡氧乙酸异辛酯(式i)
[0062]
向50ml三颈瓶中加入2-((4-氨基-3,5-二氯-6-氟吡啶-2-基)氧基)乙酸乙酯(1.6g,5.),仲辛醇(2.94g,22.6mmol),钛酸乙酯(258mg,1.)和甲苯(20ml),加热至内温110℃,反应5小时。冷却至室温,加dcm(100ml)稀释反应液,另加水(20ml
×
3)洗涤有机相,分液,无水硫酸钠干燥,浓缩得淡黄色产物1.88g,收率90.5%,hplc纯度98%。esi-ms(m/z):367.10[m+h]
。
[0063]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
技术特征:
1.一种氯氟吡氧乙酸异辛酯的制备方法,其特征在于,包括如下步骤:(1)式ii化合物在氟化试剂/相转移催化剂存在下生成式iii化合物;(2)式iii化合物与氨试剂反应生成式iv化合物;(3)式iv化合物在强碱试剂存在下,与乙醇酸乙酯反应生成式v化合物;(4)式v化合物在催化剂存在下,与仲辛醇反应生成式i化合物。2.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(1)中氟化试剂/相转移催化剂为kf/四丁基氟化铵、kf/四丁基氯化铵或均为四甲基氟化铵。3.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(1)中氟化试剂、相转移催化剂与式ii化合物的摩尔比为5~15:3~8:1。4.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(2)中的氨试剂为液氨或氨水。5.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(2)中反应温度为70℃~90℃。6.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(3)中强碱试剂为nah、碳酸铯或叔丁基锂中的一种或几种。7.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(3)中强碱试剂、乙醇酸乙酯与式iv化合物的摩尔比为1~3:1~2:1。8.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(3)中反应溶剂为dma、dmf或dmso中的一种或几种;所述反应溶剂与式iv化合物的体积/摩尔比为2~5:1。9.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(4)中催化剂为钛酸丁酯或钛酸乙酯中的一种或几种。10.根据权利要求1所述的氯氟吡氧乙酸异辛酯的制备方法,其特征在于,所述步骤(4)中催化剂、仲辛醇与式v化合物的摩尔比为0.15~0.25:2~5:1。
技术总结
本发明涉及一种氯氟吡氧乙酸异辛酯的制备方法,包括以五氯吡啶为原料,经氟代反应、氨基取代后制备得到3,5-二氯-2,6-二氟-4-氨基吡啶,再依次与乙醇酸乙酯、仲辛醇反应得到目标产物。该制备方法原料便宜易得,避免了传统工艺中大量含氯试剂的使用,安全环保;同时反应后处理简单,收率和纯度较高,适合氯氟吡氧乙酸异辛酯的工业化生产。乙酸异辛酯的工业化生产。
技术研发人员:王豪 张璞 吴耀军
受保护的技术使用者:江苏中旗科技股份有限公司
技术研发日:2021.11.26
技术公布日:2022/2/6